How to Prepare Protein Samples for Western Blot
by Fernanda Ruiz, PhD

by Fernanda Ruiz, PhD
 Western blotting is a very common and powerful technique
often used worldwide to detect, characterize and quantify proteins. Although
common, a Western blot is composed of multiple steps that require careful
consideration and planning. First, a protein sample is prepared, then subjected
to gel electrophoresis allowing the separation of native or denatured proteins
based on size. Next, the proteins are transferred onto a membrane, where an
antibody is added to bind to a specific protein, your protein of interest. This
antibody-protein complex can then be detected and imaged. Finally, you can
complete analysis of the imaged protein (Figure 1 below).
Western blotting is a very common and powerful technique
often used worldwide to detect, characterize and quantify proteins. Although
common, a Western blot is composed of multiple steps that require careful
consideration and planning. First, a protein sample is prepared, then subjected
to gel electrophoresis allowing the separation of native or denatured proteins
based on size. Next, the proteins are transferred onto a membrane, where an
antibody is added to bind to a specific protein, your protein of interest. This
antibody-protein complex can then be detected and imaged. Finally, you can
complete analysis of the imaged protein (Figure 1 below).
Click here for GoldBio’s Western Blot protocol.

Figure 1. Flowchart showing the elemental steps in Western blot.
The first step of a Western blot, the sample preparation, is of uttermost importance because the success of later steps in your immunoblotting procedure depends on the proper initial preparation of the protein, which should yield a high amount of the target protein. Western blot can be performed using purified protein samples. However, it is most common to use tissue or cell extracts, which contain a mixture of different proteins.
In the case of tissue or cell extracts, the cells must first be carefully disrupted or lysed to release the proteins without causing degradation or proteolysis. Protein samples from cell extracts, often mammalian cells grown in cell culture, are relatively easy to prepare. Preparing a protein sample from tissue might require a different strategy since tissue is more difficult to disrupt. In addition, the type of sample preparation also depends on the cellular location of the protein (cytoplasmic/soluble or membrane-bound).
It must be noted, however, that all sample preparation, whether from tissue or cells, must be performed as rapidly as possible, at a low temperature (in ice and using chilled equipment) and using the mildest buffer possible to prevent damage or degradation of proteins in the sample.
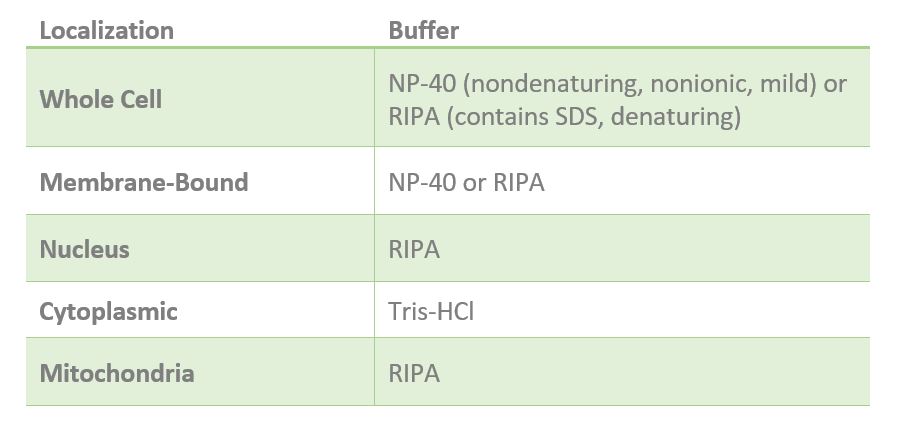
One of the most important elements of successful lysis is the lysis buffer. The lysis buffer you use largely depends on the localization of your protein of interest (Table 1). Generally, if your protein is cytoplasmic, Tris-HCl lysis buffer (20mM Tris-HCl, pH 7.5) is recommended. If your experiment requires solubilization of your protein, then a harsh buffer containing detergent can be used such as RIPA buffer. RIPA buffer can be used to isolate membrane-bound, cytosolic and nuclear proteins because it contains the denaturant sodium dodecyl sulfate (SDS), which is an ionic detergent capable of solubilizing membrane-bound proteins. This process also results in disruption of protein-protein interactions and denaturing or “unfolding” of the protein.
Alternatively, some proteins only require a mild nonionic detergent like nonidet P-40 (NP-40), which solubilizes both the plasma membrane and membranous organelles leaving the cytoskeleton intact. NP-40 is mild detergent ideal for whole cell lysates, some membrane-bound proteins and cytosolic proteins. If your protein must be extracted from the cytoskeleton (solubilized), then a harsher buffer such as RIPA may be used.
Another detergent that may be used in cell lysis is 3-cholamidopropyl dimethylammonio 1-propanesulfonate (CHAPS), which is a non-denaturing zwitterionic detergent able to solubilize membrane proteins, maintain some protein-protein interactions and disaggregate protein complexes without affecting secondary or tertiary structures. Since CHAPS can disrupt protein-protein interactions, it is not recommended for co-immunoprecipitation experiments.
Table 1. Recommended lysis buffer for proteins localized in different cell compartments.
In general, disruption of soft tissues and cell cultures can be accomplished with vigorous pipetting. However, as mentioned previously, some tissue samples: liver, muscle and skin, for example, might present a larger challenge when trying to lyse cells and extract proteins.
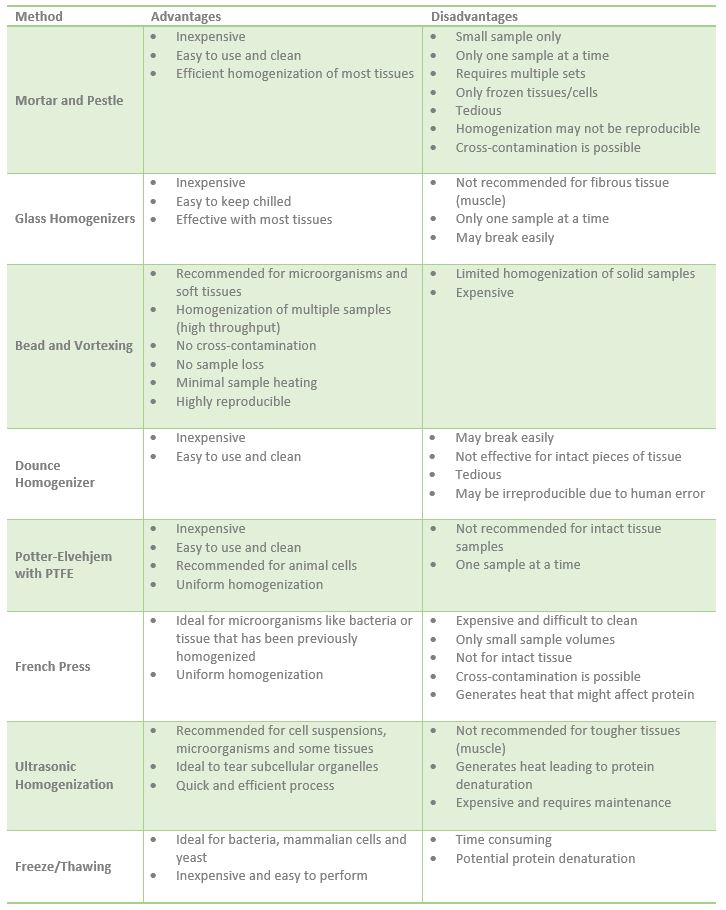
The breaking up of tissue (homogenization) and disruption of cells may require mechanical disruption in lysis buffer. Tissue can be homogenized using a French Press, glass homogenizer, sonication or mortar and pestle, etc (Table 2). The settings for each of these homogenization instruments varies depending on the type and size of tissue. Notably, the tissue must be kept cool during this homogenization step by performing on ice or using equipment that has a cooling feature.
In the case of the mortar and pestle, solid samples work best, thus, tissue is usually first frozen with liquid nitrogen. This set up is relatively inexpensive but if only a small amount of sample is available, it might be lost during grinding and result in low protein yield. Also, only one sample can be processed at a time, and must be cleaned between samples, making the whole process longer or require multiple mortar/pestle sets.
Glass homogenizers are another option when you must grind tissue. These must also be washed and decontaminated after every use and sample to prevent sample cross-contamination. These are inexpensive, easy to keep chilled and effective with most tissues. However, they may not be efficient with fibrous tissues such as muscle tissue.
Vortexing samples with beads also accomplishes shearing and homogenizing of microorganisms, mammalian cells and soft tissue like mouse brain cortex or adipose tissue. You would need the stainless steel beads and your sample in buffer. This method allows for homogenization of multiple samples and can be arranged in racks. Its disadvantages include limited homogenization of solid samples.
One very common homogenizing instrument commonly used for culture cells is the Dounce homogenizer. Unlike the grinding action of a glass homogenizer, the Dounce homogenizer shears cells leaving subcellular particles intact. This instrument is relatively inexpensive and easy to use. It can also be easily cleaned. However, it is also easily breakable and is not effective for solid pieces of tissue.
The Potter-Elvehjem with Polytetrafluorethylene (PTFE) pestle is a similar instrument to the Dounce homogenizer in that it is inexpensive, easy to use and clean, as well as not recommended for tissue homogenization. It is most commonly used for animal cells. In addition, this instrument utilizes both a shearing and grinding action to achieve homogenization.
The French Press or French Pressure Cell is an old homogenization method, usually used for microorganisms like bacteria or tissue that has been homogenized with a different instrument. Homogenates generated with the French Press are very uniform, however, it is expensive, can only handle small sample volumes and is hard to clean.
Ultrasonic homogenization is another very powerful technique that shears and disrupts cells. Sonication uses ultrasonic sounds waves created with a probe and is recommended for cell suspensions, microorganisms and some tissues. It is not recommended for tougher tissues such as muscle. One disadvantage of this method is the heat that it can generate that if not controlled might lead to protein denaturation.
Another method that has proven effective in breaking up bacteria, mammalian cells and yeast is freeze/thawing. Essentially, cells are frozen and thawed multiple times using dry ice or an ethanol bath. Freeze/thawing is very inexpensive but it can be time consuming and the multiple thawing and freezing might result in protein denaturation.
Keep in mind that plants, bacteria, yeast and fungi have cell walls that must be first disrupted before a detergent can be used to break the cell membrane and release proteins. Mechanical disruption as well as enzymes could be used to break cell walls. The cell walls of yeast, plants, fungi and bacteria can be disrupted using zymolyase, cellulases, chitinases, and lysozymes, respectively.
Table 2. Advantages and disadvantages of different homogenization equipment.
At times, the protein being studied is present at low levels or present in tissues that also express many other proteins making it difficult to detect. In this case, you can use cellular fractionation to isolate specific subcellular components and enrich for your specific protein of interest to try to achieve detection on your Western blot. Subcellular fractionation, the separation of proteins based on their physical properties, is often used to enrich cytosolic, nuclear, membrane bound and mitochondrial proteins and is performed using differential centrifugation and in some cases specific detergents. Knowing the localization and characteristics of your protein is essential since it will guide your choice of buffers as well as homogenization and centrifugation steps.
For example, membrane bound (hydrophobic) proteins may be separated from cytosolic (hydrophilic) proteins by incubating lysed cells with detergents such as Triton-X, TritonX-114, Tween-20 or digitonin among others, resulting in formation of two phases: a hydrophilic phase and a hydrophobic phase. This method can be very efficient in enriching for cytosolic and membrane proteins. However, in some cases, the high amount of detergent needed might interfere with downstream assays. Thus, you may need to remove the excess detergent (Figure 2).
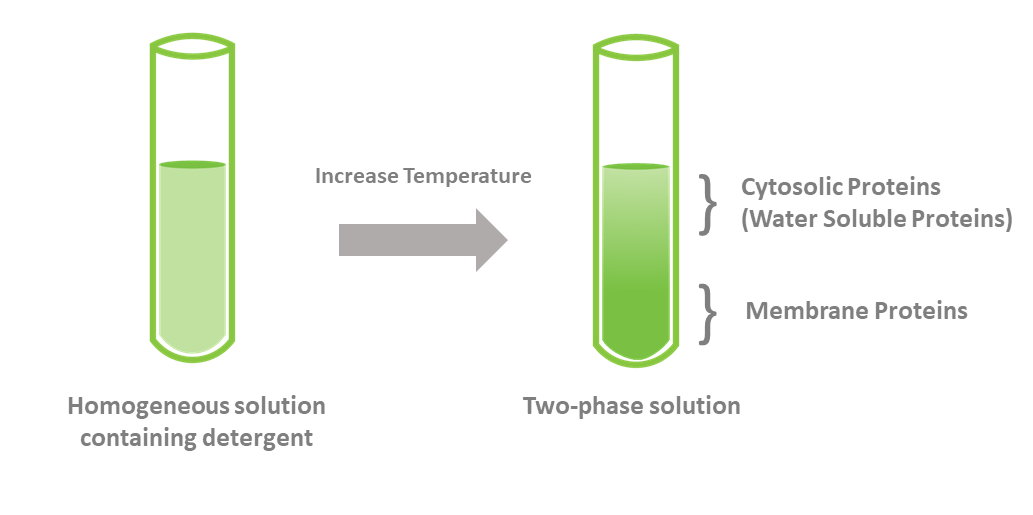
Figure 2. Separation of membrane-bound and cytosolic proteins with detergent.
In some cases, organelles--mitochondria, nuclei, and endoplasmic reticulum—can be separated using centrifugation following homogenization. This process begins by first lysing the cells by homogenization (with a Dounce homogenizer, for example) and then centrifuging the homogenate and subsequent supernatants at low speeds and then at progressively higher speeds. This method does require you to know where your protein is expressed as well as addition of protease and phosphatase inhibitors (Figure 3).
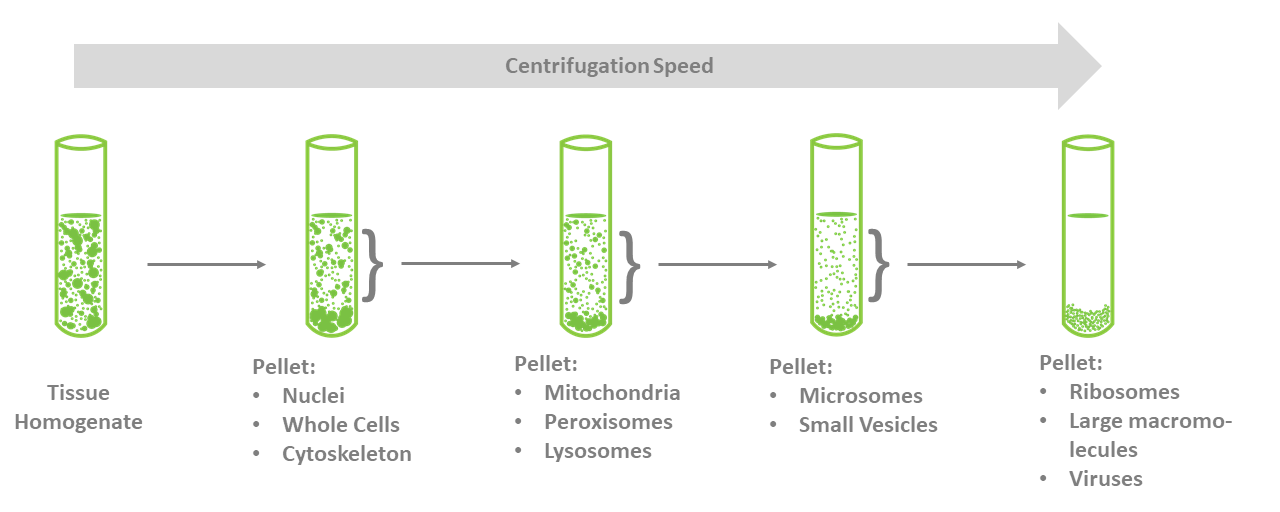 Figure 3. Subcellular fractionation.
Figure 3. Subcellular fractionation.
Tissue and cell lysis and homogenization not only release proteins, they also release proteases and phosphatases capable of degrading and altering your protein of interest. Thus, protease and phosphatase inhibitors must be used during the lysis step. Protease inhibitor cocktails are also routinely used. Below you will find a guide to different protease inhibitors and cocktails including the type of inhibitor, specificity, molecular weight, storage and recommended working concentration (Table 3). Click here for a GoldBio protease inhibitor cocktail guide.
Table 3. Guide to different protease inhibitors for protein sample preparation.
Once a protein extraction is complete, the protein concentration in each sample must be measured to be able to standardize total protein loaded onto each well of the SDS-PAGE gel. Loading equal amounts of total protein into each well ensures accurate quantification in the analysis step of your Western blot. Total protein concentration can be measured using a variety of assays including Bradford, Lowry and bicinchoninic acid (BCA) assay.
In addition, when using these different assays, a standard curve of known protein concentrations should be completed to ensure that the assay is working effectively. Then, a desired concentration must be chosen and all samples must be diluted to this concentration, making sure that protease inhibitors are also added. These samples can then be prepared for SDS-PAGE gel loading or stored at -20°C or -80°C.
Alberts B, Johnson A, Lewis J, et al. Molecular Biology of the Cell. 4th edition. New York: Garland Science; 2002. Membrane Proteins. Available from:https://www.ncbi.nlm.nih.gov/books/NBK26878/
Arnold, T., & Linke, D. (2007). Phase separation in the isolation and purification of membrane proteins. Biotechniques, 43(4), 427-440. doi:10.2144/000112566.
Bass, J. J., Wilkinson, D. J., Rankin, D., Phillips, B. E., Szewczyk, N. J., Smith, K., & Atherton, P. J. (2016). An overview of technical considerations for Western blotting applications to physiological research. Scandinavian Journal of Medicine & Science in Sports, 27(1), 4-25. doi:10.1111/sms.12702.
Clayton1, D. A. (1970, January 01). David A. Clayton. Retrieved from http://cshprotocols.cshlp.org/content/2014/10/pdb.prot080002.full
Grabski, A. C. (2009). Chapter 18 Advances in Preparation of Biological Extracts for Protein Purification. Methods in Enzymology Guide to Protein Purification, 2nd Edition, 285-303. doi:10.1016/s0076-6879(09)63018-4.
Guide to the Disruption of Biological Samples – 2012. (n.d.). Retrieved from https://opsdiagnostics.com/applications/OPSD_2012_Disruption_Guide_v1.1.pdf
Hjelmeland, L. M. (1980). A nondenaturing zwitterionic detergent for membrane biochemistry: Design and synthesis. Proceedings of the National Academy of Sciences, 77(11), 6368-6370. doi:10.1073/pnas.77.11.6368.
Holden, P., & Horton, W. A. (2009). Crude subcellular fractionation of cultured mammalian cell lines. BMC Research Notes, 2(1), 243. doi:10.1186/1756-0500-2-243.
Huber, L. A., Pfaller, K., & Vietor, I. (2003). Organelle Proteomics. Circulation Research, 92(9), 962-968. doi:10.1161/01.res.0000071748.48338.25.
Johnson, B. H., & Hecht, M. H. (1994). Recombinant Proteins Can Be Isolated from E. coli Cells by Repeated Cycles of Freezing and Thawing. Nature Biotechnology, 12(12), 1357-1360. doi:10.1038/nbt1294-1357.
Kurien, B. T., & Scofield, R. H. (2015). Western Blotting: An Introduction. Methods in Molecular Biology Western Blotting,17-30. doi:10.1007/978-1-4939-2694-7_5.
Peach, M., Marsh, N., Miskiewicz, E. I., & Macphee, D. J. (2015). Solubilization of Proteins: The Importance of Lysis Buffer Choice. Methods in Molecular Biology Western Blotting, 49-60. doi:10.1007/978-1-4939-2694-7_8.
Rodi, P., Gianello, M. B., Corregido, M., & Gennaro, A. (2014). Comparative study of the interaction of CHAPS and Triton X-100 with the erythrocyte membrane. Biochimica Et Biophysica Acta (BBA) - Biomembranes, 1838(3), 859-866. doi:https://doi.org/10.1016/j.bbamem.2013.11.006.
Fernanda Ruiz is a science content writer at Gold Biotechnology. She holds a bachelor's of science in biology from St. Mary's University and a PhD in molecular biology from Baylor College of Medicine.

Labeling antibodies with biotin or a fluorophore enables scientists to detect, track, and quantify that antibody and the molecules that it binds to. We cover...

Covalently conjugating a small molecule to an antibody’s surface is a process called antibody “labeling.” Labeling antibodies with small molecules such as biotin or fluorophores...

DNA gel electrophoresis is one of the most widely used analytical techniques in molecular biology, providing a simple and reliable way to separate DNA fragments...

Do you have a favorite restaurant that you love because you know exactly how great the experience is going to be? There are probably a...