3 Small Peptide Tags for Affinity Protein Purification
by Simon Currie, Ph.D.

by Simon Currie, Ph.D.
Affinity purification is a frequently used technique to isolate proteins for further research and other applications. However, there are a lot of different protein affinity tags, and if you have never performed affinity purification before, you might wonder which one you should use.
In this article we will discuss small peptide affinity tags, such as his-tags. These tags are a great choice because they usually do not interfere with the tagged-protein’s function, and allow versatility in where you tag the protein.
Protein affinity tags are small peptides or proteins tagged onto a target protein to facilitate its purification or detection. Three common small peptide tags are his-tags, flag-tags and strep-tags. These affinity tags have different strengths and limitations to consider when deciding which to use.
The small size of these peptide tags provides flexibility in adding them to either the amino or carboxy termini of the protein of interest – or even within a protein’s flexible loops. Since these tags are small, they usually do not impact a protein’s structure or function. However, this point should always be experimentally verified when working with a fusion protein with a new affinity tag.
In contrast, other affinity tags, such as GST-tags, consist of entire protein domains. These large tags are frequently used when the protein of interest has limited solubility in an expression host. To improve solubility, these tags need to be added to the protein’s amino terminus. To learn more about large solubility affinity tags, check out this article!
His-tag is a stretch of histidine residues that bind to nickel and other divalent transition metals. When fused to a protein, the his-tag can be used to purify that protein using a nickel column.
His-tags typically range from 6 to 10 histidine residues in length. 6x his-tags are frequently used, however lengthening the tag up to 10 histidine residues can increase the efficiency of the purification process.
These histidine residues interact with nickel and other divalent transition metal ions. Nickel ions are conjugated onto agarose beads, often via nitrilotriacetic acid (NTA), and used to bind his-tagged proteins.

Figure 1. The target his-tagged proteins (purple) bind to nickel agarose beads. Low concentrations of imidazole (blue) prevents non-target proteins from binding to the agarose beads and they are washed out.
Imidazole is the eluent for his-tags. Histidine side chains coordinate nickel through their imidazole moiety (Figures 1 and 2). Adding free imidazole in sufficient excess competes to bind with the nickel column and thereby elutes his-tags from the column. Typically, ~ 0.25 to 1 molar imidazole is used to elute his-tags from nickel columns.
Imidazole should also be added at lower concentrations to the loading and wash buffers (Figure 1). This is because many endogenous proteins contain polyhistidine stretches that act like naturally occurring his-tags and weakly bind to nickel columns (Salichs et al., 2009). In fact, depending on the protein context, even a single histidine residue can be sufficient to bind to nickel columns (Hemdan et al, 1989).

The imidazole side chains of histidine residues in the His-tag (purple) bind to Ni2+-conjugated agarose beads. Free imidazole (blue) is added in excess to elute His-tagged proteins from the Ni2+ beads.
Binding strength correlates with the length of polyhistidine residues (Fessenden, 2009; Guignet et al., 2004; Hemdan et al, 1989). However, most of these contaminating proteins can be separated from your his-tagged protein of interest by loading the protein mixture and washing with ~ 5 – 50 millimolar imidazole.
This is also why going with a longer his-tag, such as 10 histidine residues, and using a higher concentration of imidazole in the loading and wash steps can lead to higher purity after affinity purification.
His-tags are a widely used affinity purification technique, yet there are a few things to keep in mind when taking this approach.
First, the protein and buffer solutions cannot contain strong metal chelating agents such as EDTA or EGTA. This means if you are using protease inhibitors, make sure to use the EDTA/EGTA-free variety. Including EDTA/EGTA in your buffers will strip nickel off of your column thereby eliminating the binding site for your his-tagged protein!
Secondly, his-tag purification may not be the best approach for proteins that have metal cofactors because these cofactors will bind to the agarose beads in the nickel column. In some cases, this limitation can be overcome by including excess metal in your purification buffers. However, if you know that your protein has a key metal cofactor, it may be worth considering other affinity tag approaches, such as those discussed below.
A flag-tag is the small hydrophilic peptide with the sequence DYKDDDK. It was called a flag-tag because it is used as a marker, or a flag, to detect proteins that it is added onto (Hopp et al., 1988).
Antibodies raised to recognize the flag tag are used to purify and detect flag-tagged proteins.
The flag-tag sequence was rationally designed to overcome several limitations of fusion protein tags prior to its invention including:
The DYKDDDK tag solved these problems because its short, hydrophilic sequence is well tolerated by most proteins.
If anything, the flag-tag may increase protein solubility and expression in certain expression host species (Schuster et al., 2000). Also, the tag can be eluted and cleaved under mild, physiological conditions that will not denature the protein of interest.
Many antibodies have been developed to recognize flag-tag. The 3 main ones used today are: M1, M2, and M5. Each antibody has different strengths and limitations in detecting and purifying flag-tagged proteins (Table 1).
We will describe flag-tagged protein purification using M2, since this antibody allows the most flexibility in tag location and for mild, physiological elution conditions (Einhauer and Jungbauer, 2001). Purification with either M1 or M5 would be very similar with altered elution conditions (Table 1).
Table 1. Antibodies used to recognize flag tags.
| Antibody | Elution | Notes |
|---|---|---|
| M1 | Depletion of Ca2+ from buffer | M1-flag interaction is Ca2+-dependent Only recognizes N-terminal flag tags |
| M2 | Addition of free flag peptide or Acidic pH |
Recognizes N- or C-terminal flag tags |
| M5 | Addition of free flag peptide or Acidic pH |
Higher affinity for N-terminal flag tags |
Cell lysate, or another complex biochemical mixture, containing a flag-tagged protein would be loaded onto a column with M2 antibodies conjugated to agarose beads. After washing the column, the flag-tagged protein would then be eluted by adding excess flag peptide to disrupt the interaction between the flag-tagged protein and the M2-labelled beads.
You probably noticed that this is the same bind-wash-elute process described for his-tags and Ni2+ columns (Figures 1 and 2), the molecular identities of the binding and eluting agents are just different. See below for a more general description of the purification process used by all 3 of these affinity tags.
If not using the flag-tag to detect the protein in downstream applications, it can be cleaved using enterokinase. For proteins tagged on the amino termini, enterokinase will leave a “scarless” cleavage – that is, no amino acids from the flag tag will remain on the protein of interest (Figure 3).
Since its design, there have been several modifications for using the flag-tag. Two of the more useful modifications are:
Flag tags and flag-recognizing antibodies, in all of their forms, are a popular choice of tag and are widely used for the detection and purification of proteins.

Figure 3. An added protease cleavage site is not necessary for flag-tags. Enterokinase (depicted as a pair of scissors) cuts after the flag-tag leaving the protein of interest without any additional amino acids.
A Strep-tag is a short peptide - WRHPQFGG - that binds to the bacterial protein streptavidin with extremely high affinity. Streptavidin also binds to biotinylated proteins using the same protein surface (Figure 4), so strep-tagged proteins can be eluted from streptavidin with biotin.
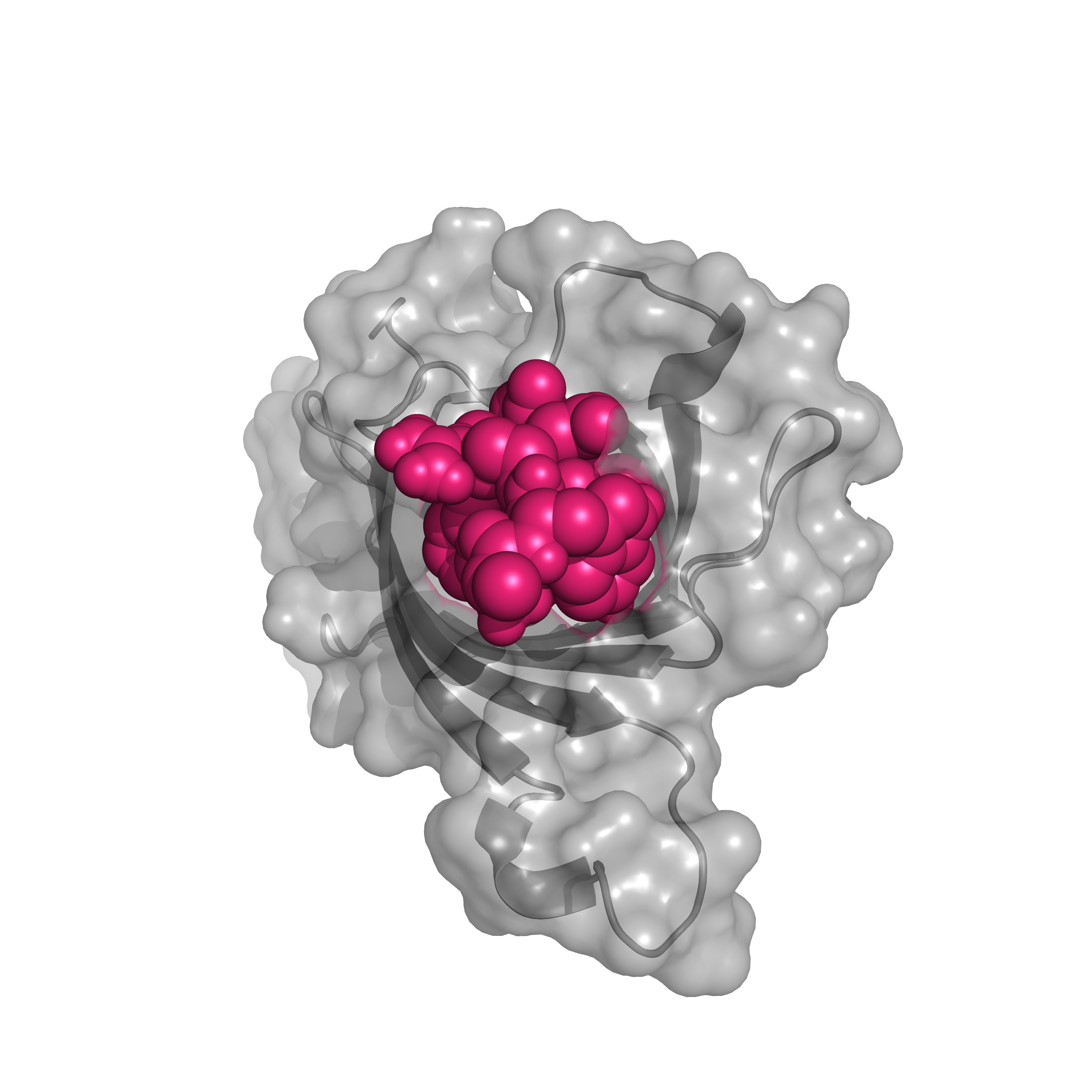
Figure 4. Strep-tag (pink) binding to streptavidin (gray) (PDB: 1RST).
The original Strep-tag was selected from a genetic screen searching for small peptides that bind to streptavidin. The high affinity of this interaction means that using a Strep-tag for affinity purification results in very little contamination with other proteins.
Strep-tagged proteins are loaded onto a column with streptavidin-conjugated agarose beads. After washing the beads, an excess of biotin, or one of its derivatives such as desthiobiotin, is used to elute the strep-tagged protein.
Since the Strep-tag’s initial report in 1993, several advances have been made to the tag.
The original strep-tag could only be fused on the carboxy terminus of the protein of interest. Strep-tag II - WSHPQFEK - allows greater flexibility and can be added to either the amino or carboxy termini of the fusion protein.
Additionally, Strep-Tactin, a variant of streptavidin with improved binding capacity to Strep-tag II was engineered.
These advances make the Strep-tag II / Strep-Tactin combination a popular choice for affinity purification, particularly when only one purification step is going to be performed.
Small peptide affinity tags are a great tool to add onto your protein of interest to aid in purifying your protein, and for detecting your protein in experiments and assays.
First let’s cover the general purification strategy for small peptide affinity tags.
A complex biochemical mixture, such as cell lysate in which the protein has been overexpressed, is loaded onto a column that will bind to the affinity tag. This column will have an interacting partner molecule - such as metal ions, small molecules, or proteins - conjugated onto agarose beads (Figure 5).
The affinity tagged protein will bind to the column, whereas other cellular biomolecules will flow through the column. A wash step, or multiple wash steps, can be used here to ensure molecules without the affinity tag do not remain bound to the column.
Then the tagged protein can be eluted from the column by adding a molecule that competes with the tag-column interaction into the elution buffer.
Alternatively, buffer conditions can be altered to weaken the tag-column interaction, such as by changing the pH.
Lastly, if there is a protease cleavage site between the tag and the protein of interest (Figure 3), the protease that recognizes that site can be added and incubated with the column before washing the cleaved protein off.
This general purification strategy is applicable for the purification of his-, flag-, and strep-tagged proteins. See each section below for further details, tips, and tricks for the execution of each specific type of affinity purification.

Figure 5. Purification of affinity-tagged proteins. Affinity-tagged proteins bind to agarose beads conjugated with interacting partner molecule (column 2). After washing, tagged-proteins are eluted by adding an elution buffer that weakens the interaction between the tag and the liganded bead (column 3).
Affinity tags are commonly used in protein purification, and they are incredibly user-friendly. However, to successfully purify your proteins and ensure there isn’t conflict with any of your downstream applications, there are 2 important considerations.
Small peptide affinity tags are typically innocuous when added onto a protein of interest, and they do not impact that protein’s function or structure. As such, small peptide tags can typically be added to either the amino or carboxy termini of a protein.
However, it is worth verifying that the protein’s function is not perturbed by the tag. If the tag does interfere with protein function, it may mean that the tag disrupts folding of the protein, or that it binds to an important interface on the protein’s surface. This is where the versatility of short peptide affinity tags is an excellent feature!
In these cases move the tag to the other side and retesting
the protein’s function (Figure 6). This change will likely fix any issues
caused by the tag.

Figure 6. Small peptide tags can usually be added to the amino- or the carboxy termini without influencing a protein’s function and structure. It is important to verify that the tag is inert, and if not move the tag to the other side.
When designing an expression construct for fusion proteins, you’ll want to ask yourself if the affinity tag is desirable or tolerable for your downstream uses.

Figure 7. For applications where it is desirable to leave the affinity tag on, no protease cleavage site is needed (top). However, if the affinity tag is only being used during purification and is not needed in downstream applications, a protease cleavage site can be added between the affinity tag and protein of interest.
For example, tagged proteins are required in several assays that measure molecular interactions, such as surface plasmon resonance or biolayer interferometry. When purifying proteins for these applications, you would want to retain the affinity tag on the protein of interest.
If it is not necessary to keep the affinity tag for your downstream uses, you may consider adding a protease cleavage site between the affinity tag and your protein of interest (Figure 7). The flag-tag protein sequence already contains a protease cleavage site within the affinity tag, but many other tags such as a his-tag and a strep-tag would need a cleavage site to be added.
Each of these small peptide affinity tags has its own strengths and limitations that are worth carefully considering before deciding which tag to use for purification or detection of your protein (Table 2).
Table 2. Advantages and limitations of different affinity tags.
| Tag | Advantages | Limitations |
|---|---|---|
| His-tag | Amino or Carboxy termini tagging Amenable to many downstream applications |
Relatively high contamination with cellular proteins |
| Flag-tag | Protease cleavage site included Amino or Carboxy termini tagging |
|
| Strep-tag | High affinity and low contamination | Only Carboxy terminus tagging |
| Strep-tag II | High affinity and low contamination Amino or Carboxy termini tagging |
In addition, while his-, flag-, and strep-tags are 3 of the most frequently used affinity tags, there are additional tags that work using similar principles including: HA-, Myc-, T7-, and V5-tags.
With a little trial and error, you are sure to find a small peptide affinity tag suitable for your protein of interest and desired uses!

In the story of cell culture, Mycoplasma is the clear antagonist. These tiny, malleable bacteria lack a rigid cell wall and are notoriously difficult to...

When doing cell culture work, the term Mycoplasma has probably popped up. Mycoplasma is a genus of bacteria that lacks a cell wall and is...

Antibiotics and cell selection agents are used to select for specific cell populations to, for instance, generate a stable cell line or conduct a genetic...

Antibiotics are powerful tools that protect cell culture from contamination and help select for transfected cells. However, antibiotics can also have subtle impacts on resistant...