Breakthroughs in Computational Protein Design
by Simon Currie, Ph.D.

by Simon Currie, Ph.D.
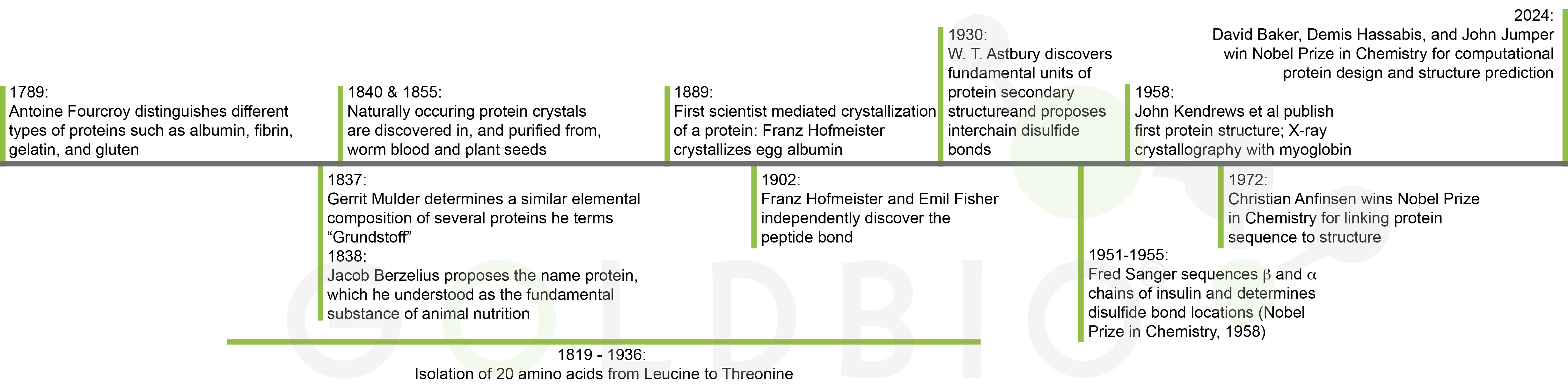
The history of protein and protein purification research
stretches back more than 200 years and includes several Nobel prizes (Figure
1). Modern-day scientists use the wealth of information and techniques created
by past researchers to design protein constructs, purify them, and test them
for activity, binding, or solve their structure.
And now, this rich past paired with new technology is giving us cutting-edge research, informing novel designer proteins and interrogating more proteins than ever before. And, it may lead us into a future where the design, make, test, analyze cycle is completely automated.
Imagine cutting years of labor-intensive research down to a couple weeks - catalyzing new discoveries for medical, environmental, and other applications.
To really understand the breakthroughs that are enabling us to explore protein research and make designer proteins that serve as custom-fit solutions, we need to step back and truly understand the vastness of protein sequence space.
What proteins will be purified in the future?
Infinitesimally small sampling of protein sequence space
AI advances in computational protein prediction
Supporting basic scientific research
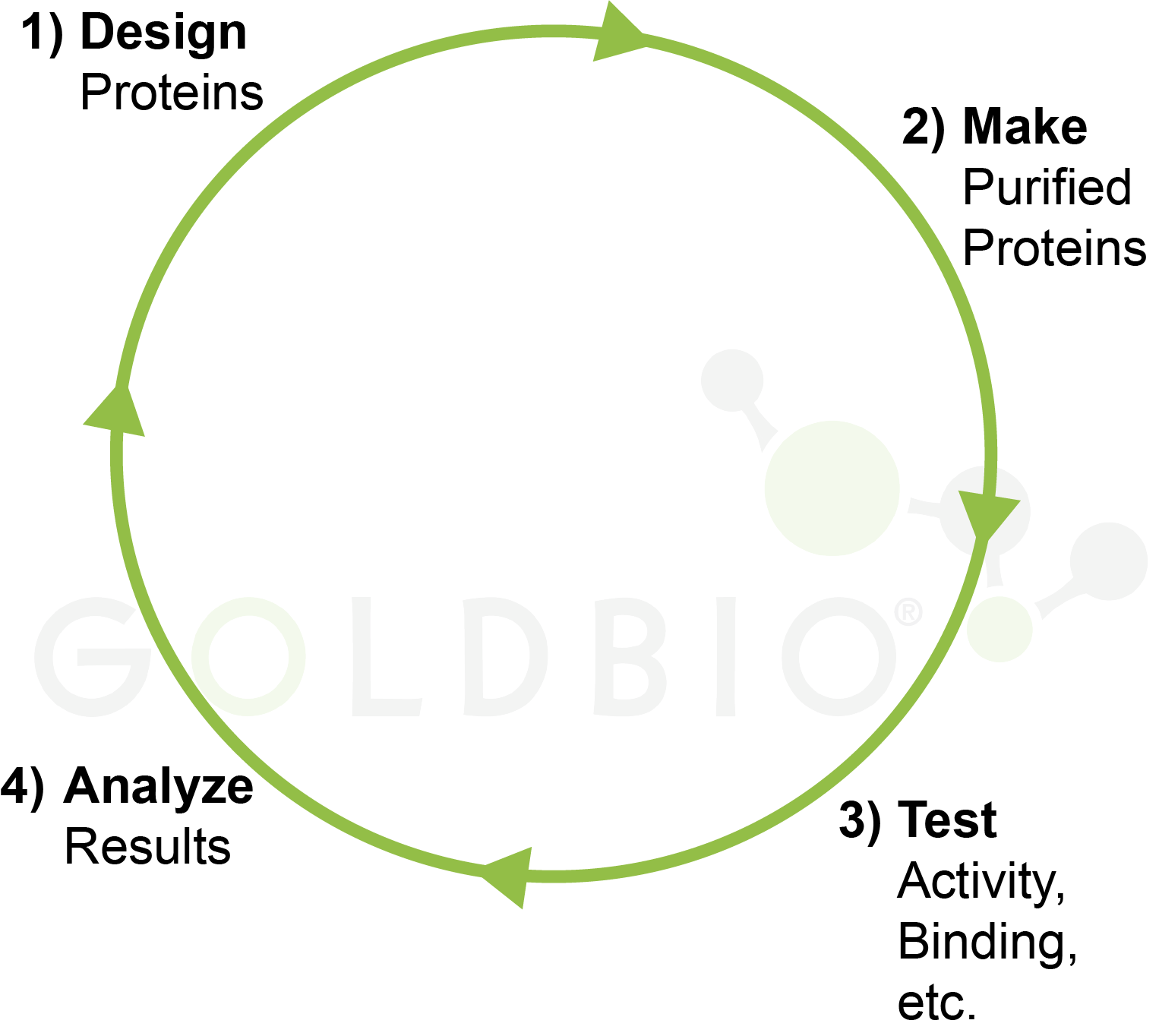
Figure 2. Design,
make, test, analyze cycle in biomedical research.
Most proteins that have been investigated are those found in nature, or closely related to natural proteins. However, this collection represents a tiny fraction of total possible proteins.
Recent advances in the computational prediction of proteins are enabling us to venture out into the universe of all possible proteins and use novel designer proteins as medicines, environmental janitors, and more.
To really understand how meaningful recent technological advances are to protein research, we need to keep in mind that scientists have explored only an infinitesimal fraction of the potential protein sequence space.
Potential protein sequence space is the collection of all proteins that could be made using unique strings of amino acids.
We can think of this like a really big combination lock, where one of the twenty amino acids can be chosen in each of the different spaces. However, while most combination locks only have 3 or 4 spaces, proteins are typically a few hundred amino acids long, and the longest protein discovered so far is over seventy thousand amino acids in length (Fallon et al, 2024; Nevers et al, 2023). That would be an enormous number of combinations for the lock!
Given the magnitude of all possible protein sequence combinations, conceptually we could really consider the fraction of explored proteins to just be zero. Of course, there are lots of proteins out there, so the protein space we’ve sampled isn’t actually zero, it’s just really, really small.
The paucity of sampled protein space is hard to grasp because there are a lot of proteins that we know about. For example, there are a little over 20,000 protein-encoding genes in the human genome (Salzberg, 2018). If we expand past humans and consider all known proteins from any organism, then we’re up to around 200 million proteins (Jumper et al, 2021).
So how big does the denominator of possible proteins have to be to completely swamp the numerator of 200 million known proteins? It’s really big! For a protein that is N amino acids long, the number of unique proteins that long is calculated by 20N. This is because there are 20 natural amino acids that can go into each position. So, you multiply 20 x 20 x 20 up to N times to get the total possible number of unique amino acid sequences (Figure 3).

Figure 3. The
number of unique sequences for a protein that is N amino acids long is 20
N
if considering only the 20 canonical amino acids.
The amount of protein sequence space that we’ve explored is much, much smaller than 1 x 10-300. In decimal form that is less than:
0.00000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000
0000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000
000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000001
Again, this number is so small it is really hard to conceptualize. But, if the possible protein space was the size of our observable universe, then the protein space that nature has sampled would be much smaller than the size of a single electron.
if the possible protein space was the size of our observable universe, then the protein space that nature has sampled would be much smaller than the size of a single electron.

Imagine trying to
find a single, specific electron among all the stars.
Given these astronomical numbers, you can appreciate that we are nowhere close to experimentally generating all of the possible proteins that could exist. That number is simply too large! And even with all the advances in automated protein purification, we’ll still likely never get close to exploring all of the possible protein sequence space.
Let’s investigate a concrete example to help reinforce this rather abstract concept. Traditionally, many designer proteins are very similar to their natural starting point. By designer proteins, I mean proteins that do not exist in nature, but were designed for a specific structure or function.
Take insulin, for example; there are new insulin analogs that increase its stability, allowing patients to dose less frequently and on a more convenient schedule (Tiwari et al, 2024). These insulin analogs are very similar to natural insulin, however, but with just a few tweaks around the edges to modify its properties.
So, the question is, is there another completely-different protein out there that could do insulin’s job as good or better than the natural protein and all of its current analogs? Given the astronomical size of total protein sequence space, the answer is likely yes. But where would we start looking?
To revisit our universe analogy, most designer proteins have been within the same city as the natural proteins from which they were derived. And for most of this history, we haven’t had the tools to explore the universe of alternative protein solutions, or to know where to start looking.
The good news is that we really don’t need to explore the entire universe of protein sequence space because advanced computational methods are helping home in on alternative starting points.
Let me make a quick point here to explicitly discuss the structure-function relationship for proteins. For natural proteins, it’s canonically stated that structure will determine their function. By knowing what domains are in a protein, you have a good starting point for understanding if the protein is an enzyme, what other types of biomolecules it interacts with, etc.
What’s new is that we can now design the structure of novel designer proteins with a specific function in mind. Most protein designer algorithms work within this structure-function paradigm, and search for a specific protein structure that should provide the desired function (Baek et al, 2021; Jumper et al, 2021). However, there are also a few groups searching for protein function in a structure agnostic fashion (Figure 4) (Rives et al, 2021).
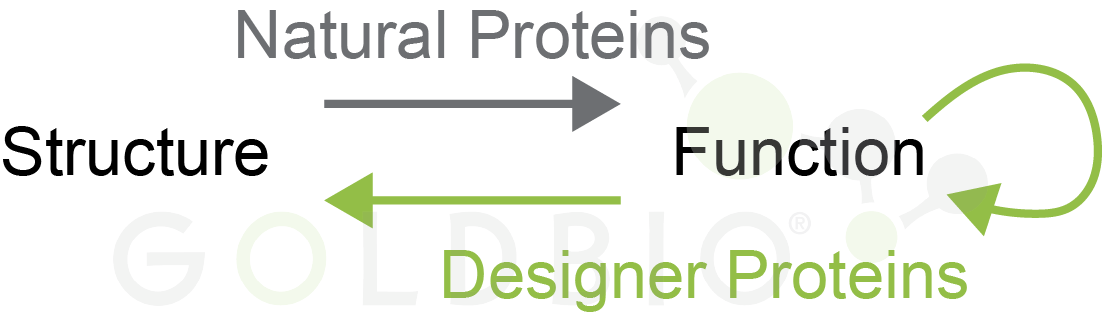
Figure 4.
Natural proteins (gray) have a structure that defines their function. Designer
proteins are made for a specific function, either by finding a structure for
that function, or in a structure agnostic fashion.
For example, let’s say you wanted to design an enzyme. You could use structure predictions to ask the question “what are all the protein scaffolds that can host an active site conducive for catalyzing this reaction?” Then you would optimize around these structures to figure out which had your desired activity profile.
Alternatively, there are algorithms that will optimize the activity itself, without specifically considering the structure. In the end, any of these active proteins will still result in a structure that facilitates the enzyme’s active site.
A lot of the work within the structure-function paradigm is enabled by impressive advances in protein structure prediction. By and large, if you pick a random protein sequence that no one has ever explored before, algorithms can predict what that protein will look like with pretty good accuracy.
This means that we can now explore the much broader universe of protein space and find the ones that work for various designer applications, instead of being limited to the neighborhood of natural protein sequences.
The Nobel Prize in Chemistry in 2024 was awarded to Drs. David Baker, Demis Hassabis, and John Jumper for their immense contributions enabling protein structure prediction (The Royal Swedish Academy of Sciences, 2024).
Why is it so useful to be able to predict protein structures? These structures are transforming the way we do basic scientific research, and have opened the floodgates for protein-based applications in a variety of fields.
In basic biochemistry and molecular biology research, we often want to know what proteins look like. This information helps us assess what their functions are, how they interact with partner molecules, and more. And structure is crucial for understanding the role of proteins in normal physiology, as well as diseased states.
Experimental protein structure determination is often a long an arduous process. From cloning an expression construct, to purifying the protein, and finally solving its structure; this process often involves many rounds of troubleshooting and can take months, years, or even decades to get the final protein structure. I should note that experimental protein structures were foundational in training the AI models that now predict protein structures as they learned on the hundreds of thousands of protein structures that scientists had already produced experimentally (Berman et al, 2000).
Even with these new powerful structure prediction tools, there are still instances where you need to get an actual, experimental structure. For example, if you’re developing a new drug that works by inhibiting a target protein, it would be crucial to know exactly how that drug binds to the protein. A prediction could help, but you’ll want to verify that the prediction is accurate.
In other cases, the structure might not be absolutely necessary, and there are other ways that you could validate the predicted structure besides actually getting the experimental structure itself.
For example, if you’re studying a protein-protein interaction, you could use AlphaFold to generate a prediction for what that interaction looks like. There may be additional data that helps you analyze the likelihood of that prediction such as disease-associated mutations that cluster around that predicted interaction. In this case, you may decide that the predicted structure is good enough or to validate the prediction biochemically with point mutations in either of the proteins (van Breugel et al, 2022).
Let’s consider again those astronomical numbers we discussed above in terms of sampling the potential protein sequence space. The numerator shouldn’t be underestimated. Look all around you at the miracle of life. 200 million proteins, apparently, is sufficient to power the myriad diversity of life – humans, animals, plants, fungi, bacteria, and all the unique things these species do to survive: photosynthesis, metabolism, outrun tigers, design skyscrapers, etc.
The good thing about there being so many additional possible proteins is that we are just scratching the surface of these proteins’ potential. That means there is a great chance that we can design novel proteins to solve pressing current problems in fields such as medicine, materials, and the environment.
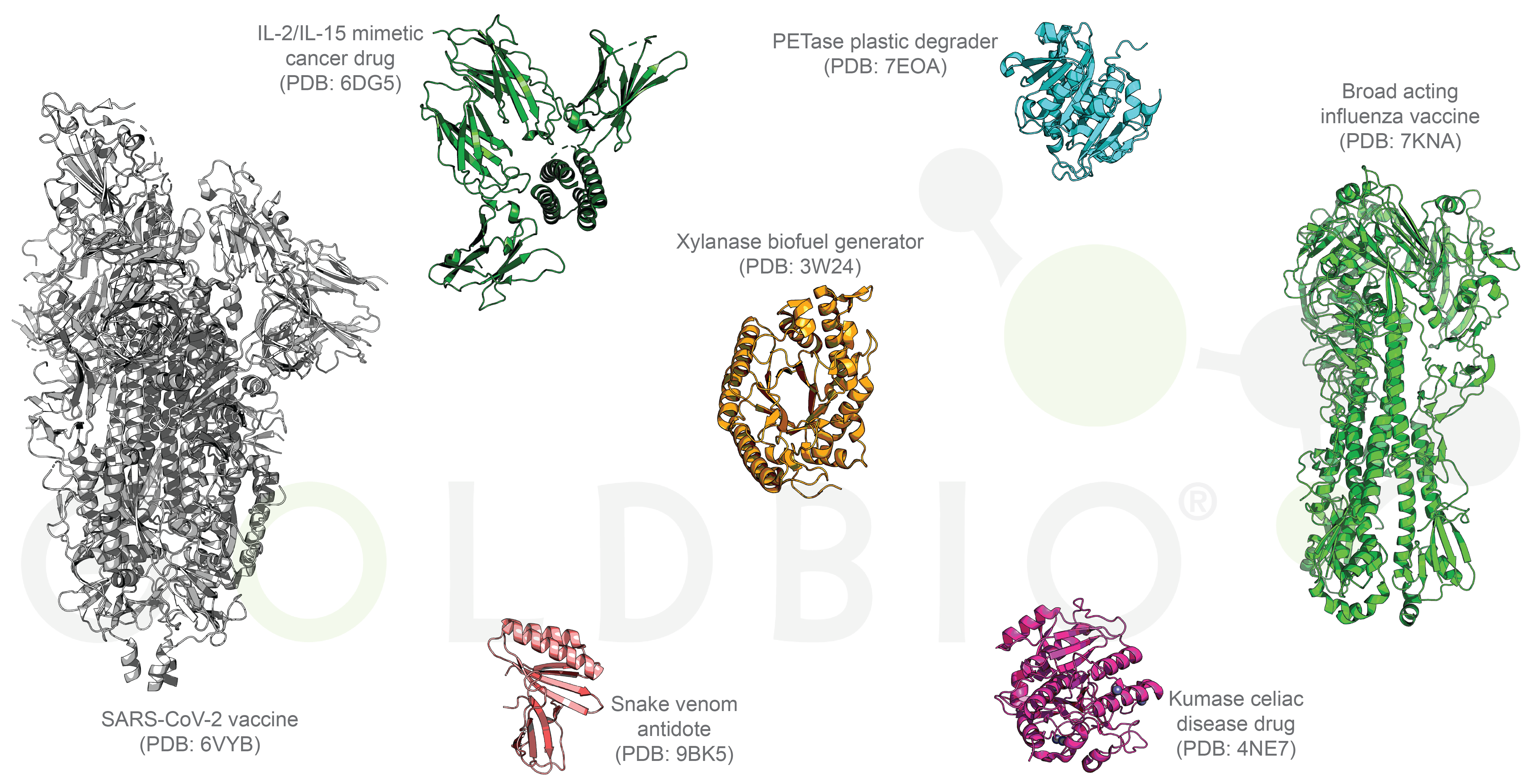
Figure 5. Compilation of designer protein structures.
What are some of these problems that designer proteins can help solve? In medicine, novel proteins are broad-acting flu and Covid-19 vaccines, drugs for cancer and celiac disease, drug delivery systems, and an antidote for snake venom (Figure 5) (Boyoglu-Barnum et al, 2021; Silva et al, 2019; Vázquez Torres et al, 2025; Walls et al, 2020; Wolf et al, 2015; Yang et al, 2024).
Designer proteins are cleaning up our environment by sequestering toxic metals, degrading plastic waste, converting agricultural waste into biofuels, and capturing greenhouse gases (Cui et al, 2024; Lipsh-Sokolik et al, 2023; Zhang et al, 2025). See Table 1 for a non-exhaustive list of companies actively developing designer protein applications for these purposes.
Table 1. Companies working on designer proteins
|
Company |
Applications |
|
Cradle Bio |
Therapeutics, Diagnostics, Food, Chemicals, Agriculture |
|
Generate Biomedicines |
Therapeutics |
|
Latent Labs |
Therapeutics |
|
Arzeda |
Therapeutics, Materials, Food, Household Goods |
|
Cycuria Therapeutics |
Drug Delivery |
|
Glox Therapeutics |
Antibiotics |
|
Monod Bio |
Clinical Diagnostics, Research Tools |
|
Outpace Bio |
Immunotherapies |
|
Cyrus Biotech |
Autoimmune Biologics |
So, that is how advances in computational predictions of proteins are enabling scientists to explore the vast protein sequence space to design novel applications for protein therapeutics, environmental cleaners, and more.
Baek, M., DiMaio, F., Anishchenko, I., Dauparas, J., Ovchinnikov, S., Lee, G. R., Wang, J., Cong, Q., Kinch, L. N., Schaeffer, R. D., Millán, C., Park, H., Adams, C., Glassman, C. R., DeGiovanni, A., Pereira, J. H., Rodrigues, A. V., van Dijk, A. A., Ebrecht, A. C., Opperman, D. J., … Baker, D. (2021). Accurate prediction of protein structures and interactions using a three-track neural network. Science (New York, N.Y.), 373(6557), 871–876. https://doi.org/10.1126/science.abj8754
Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N., & Bourne, P. E. (2000). The Protein Data Bank. Nucleic acids research, 28(1), 235–242. https://doi.org/10.1093/nar/28.1.235
Boyoglu-Barnum, S., Ellis, D., Gillespie, R. A., Hutchinson, G. B., Park, Y. J., Moin, S. M., Acton, O. J., Ravichandran, R., Murphy, M., Pettie, D., Matheson, N., Carter, L., Creanga, A., Watson, M. J., Kephart, S., Ataca, S., Vaile, J. R., Ueda, G., Crank, M. C., Stewart, L., … Kanekiyo, M. (2021). Quadrivalent influenza nanoparticle vaccines induce broad protection. Nature, 592(7855), 623–628. https://doi.org/10.1038/s41586-021-03365-x
van Breugel, M., Rosa E Silva, I., & Andreeva, A. (2022). Structural validation and assessment of AlphaFold2 predictions for centrosomal and centriolar proteins and their complexes. Communications biology, 5(1), 312. https://doi.org/10.1038/s42003-022-03269-0
Cui, Y., Chen, Y., Sun, J., Zhu, T., Pang, H., Li, C., Geng, W. C., & Wu, B. (2024). Computational redesign of a hydrolase for nearly complete PET depolymerization at industrially relevant high-solids loading. Nature communications, 15(1), 1417. https://doi.org/10.1038/s41467-024-45662-9
Fallon, T. R., Shende, V. V., Wierzbicki, I. H., Pendleton, A. L., Watervoort, N. F., Auber, R. P., Gonzalez, D. J., Wisecaver, J. H., & Moore, B. S. (2024). Giant polyketide synthase enzymes in the biosynthesis of giant marine polyether toxins. Science (New York, N.Y.), 385(6709), 671–678. https://doi.org/10.1126/science.ado3290
Ingraham, J. B., Baranov, M., Costello, Z., Barber, K. W., Wang, W., Ismail, A., Frappier, V., Lord, D. M., Ng-Thow-Hing, C., Van Vlack, E. R., Tie, S., Xue, V., Cowles, S. C., Leung, A., Rodrigues, J. V., Morales-Perez, C. L., Ayoub, A. M., Green, R., Puentes, K., Oplinger, F., … Grigoryan, G. (2023). Illuminating protein space with a programmable generative model. Nature, 623(7989), 1070–1078. https://doi.org/10.1038/s41586-023-06728-8
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., Tunyasuvunakool, K., Bates, R., Žídek, A., Potapenko, A., Bridgland, A., Meyer, C., Kohl, S. A. A., Ballard, A. J., Cowie, A., Romera-Paredes, B., Nikolov, S., Jain, R., Adler, J., Back, T., … Hassabis, D. (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596(7873), 583–589. https://doi.org/10.1038/s41586-021-03819-2
Lipsh-Sokolik, R., Khersonsky, O., Schröder, S. P., de Boer, C., Hoch, S. Y., Davies, G. J., Overkleeft, H. S., & Fleishman, S. J. (2023). Combinatorial assembly and design of enzymes. Science (New York, N.Y.), 379(6628), 195–201. https://doi.org/10.1126/science.ade9434
Martz, E. (April, 2002). Timeline of Protein Chemistry. https://www.umass.edu/microbio/chime/pe_beta/pe/protexpl/histprot.htm
Nevers, Y., Glover, N. M., Dessimoz, C., & Lecompte, O. (2023). Protein length distribution is remarkably uniform across the tree of life. Genome biology, 24(1), 135. https://doi.org/10.1186/s13059-023-02973-2
Prabhune, M. (2024, October 17). The Journey to a Nobel Prize: A Protein Design and Structure Research Timeline. The Scientist. https://www.the-scientist.com/the-journey-to-a-nobel-prize-a-protein-design-and-structure-research-timeline-72256
Rives, A., Meier, J., Sercu, T., Goyal, S., Lin, Z., Liu, J., Guo, D., Ott, M., Zitnick, C. L., Ma, J., & Fergus, R. (2021). Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proceedings of the National Academy of Sciences of the United States of America, 118(15), e2016239118. https://doi.org/10.1073/pnas.2016239118
Salzberg S. L. (2018). Open questions: How many genes do we have?. BMC biology, 16(1), 94. https://doi.org/10.1186/s12915-018-0564-x
Silva, D. A., Yu, S., Ulge, U. Y., Spangler, J. B., Jude, K. M., Labão-Almeida, C., Ali, L. R., Quijano-Rubio, A., Ruterbusch, M., Leung, I., Biary, T., Crowley, S. J., Marcos, E., Walkey, C. D., Weitzner, B. D., Pardo-Avila, F., Castellanos, J., Carter, L., Stewart, L., Riddell, S. R., … Baker, D. (2019). De novo design of potent and selective mimics of IL-2 and IL-15. Nature, 565(7738), 186–191. https://doi.org/10.1038/s41586-018-0830-7
The Royal Swedish Academy of Sciences. (2024, October 9). The Nobel Prize in Chemistry 2024. https://www.nobelprize.org/prizes/chemistry/2024/press-release/
Tiwari, D. D., Thorat, V. M., & Pakale, D. P. V. (2024). Newer Insulin Preparations and Insulin Analogs. Cureus, 16(11), e74593. https://doi.org/10.7759/cureus.74593 Tiwari, D. D., Thorat, V. M., & Pakale, D. P. V. (2024). Newer Insulin Preparations and Insulin Analogs. Cureus, 16(11), e74593. https://doi.org/10.7759/cureus.74593
Vázquez Torres, S., Benard Valle, M., Mackessy, S. P., Menzies, S. K., Casewell, N. R., Ahmadi, S., Burlet, N. J., Muratspahić, E., Sappington, I., Overath, M. D., Rivera-de-Torre, E., Ledergerber, J., Laustsen, A. H., Boddum, K., Bera, A. K., Kang, A., Brackenbrough, E., Cardoso, I. A., Crittenden, E. P., Edge, R. J., … Baker, D. (2025). De novo designed proteins neutralize lethal snake venom toxins. Nature, 639(8053), 225–231. https://doi.org/10.1038/s41586-024-08393-x
Walls, A. C., Fiala, B., Schäfer, A., Wrenn, S., Pham, M. N., Murphy, M., Tse, L. V., Shehata, L., O'Connor, M. A., Chen, C., Navarro, M. J., Miranda, M. C., Pettie, D., Ravichandran, R., Kraft, J. C., Ogohara, C., Palser, A., Chalk, S., Lee, E. C., Guerriero, K., … King, N. P. (2020). Elicitation of Potent Neutralizing Antibody Responses by Designed Protein Nanoparticle Vaccines for SARS-CoV-2. Cell, 183(5), 1367–1382.e17. https://doi.org/10.1016/j.cell.2020.10.043
Wolf, C., Siegel, J. B., Tinberg, C., Camarca, A., Gianfrani, C., Paski, S., Guan, R., Montelione, G., Baker, D., & Pultz, I. S. (2015). Engineering of Kuma030: A Gliadin Peptidase That Rapidly Degrades Immunogenic Gliadin Peptides in Gastric Conditions. Journal of the American Chemical Society, 137(40), 13106–13113. https://doi.org/10.1021/jacs.5b08325
Yang, E. C., Divine, R., Miranda, M. C., Borst, A. J., Sheffler, W., Zhang, J. Z., Decarreau, J., Saragovi, A., Abedi, M., Goldbach, N., Ahlrichs, M., Dobbins, C., Hand, A., Cheng, S., Lamb, M., Levine, P. M., Chan, S., Skotheim, R., Fallas, J., Ueda, G., … Baker, D. (2024). Computational design of non-porous pH-responsive antibody nanoparticles. Nature structural & molecular biology, 31(9), 1404–1412. https://doi.org/10.1038/s41594-024-01288-5
Zhang, Y., Srijay, D., Quinn, Z., Chatterjee, P. (2025) Metalorian: De Novo Generation of Heavy Metal-Binding Peptides with Classifier-Guided Diffusion Sampling. bioRxiv. https://doi.org/10.1101/2025.07.10.664242

Antibiotics and cell selection agents are used to select for specific cell populations to, for instance, generate a stable cell line or conduct a genetic...

Antibiotics are powerful tools that protect cell culture from contamination and help select for transfected cells. However, antibiotics can also have subtle impacts on resistant...

Antibiotics and cell selection agents are used to isolate cells that contain a particular resistance marker from a mixed population. These powerful reagents are used...

During my undergraduate internship I was making different formulations of insulin nanoparticles that would, in theory, be delivered through an inhaler and into the lungs....